![]()
![]()
1) Definition der
Tay-Sachs-Krankheit*:
* Der Name geht
zurück auf die Erstbeschreibung durch
Bernhard Sachs 1889
(1858-1944, amerikanischer Neurologe)
und Warren Tay 1881
(1853-1927, britischer Ophtalmologe).
Die
Tay-Sachs-Krankheit ist eine erbliche Fettspeicherkrankheit,
die bei Kindern in der Regel zwischen dem 3. und 7. Lebensmonat auftritt.
Sie gehört zu den Gangliosidosen, einer Gruppe von erblich bedingten
Stoffwechselerkrankungen, die man
auch als "lysosomale*
Speicherkrankheiten" bezeichnet.
*Lysosomen sind kleine Zellorganellen
die eine Vielzahl von Hydrolasen
(=Enzyme) enthalten, welche den Abbau von Nucleinsäuren (DNA und RNA),
Proteinen, Lipiden und Kohlenhydraten aber auch defekten Zellbestandteilen
katalysieren. Weiterhin sind sie am Immunsystem beteiligt und erfüllen
somit bestimmte Abwehrfunktionen wie z.B. die Beseitigung von Bakterien
und anderer körperfremden Stoffen.
Man kann sie sozusagen als „Organellen der Abfallbeseitigung“ betrachten.
Bei einer lysosomalen Speicherkrankheit werden Stoffe aufgrund eines
Enzymmangels nicht weiterverarbeitet, so dass sie sich in den Lysosomen
ablagern. Im Falle der Tay-Sachs-Krankheit ist die Ursache ein Mangel
an Hexoseaminidase A - dieses Enzym leistet den Ab- bzw. Umbau von
Sphingolipiden*- wodurch eine Anhäufung von Lipidmengen vor allem im
Gehirn erfolgt. Durch diese Fettansammlungen kommt es zum Absterben
von Nervenzellen (Neuronen), deshalb zählt die Tay-Sachs-Krankheit zu
den sogenannten „neurodegenerativen Krankheiten“.
*Sphingolipide:
eine Gruppe von Lipiden, die am Aufbau
von Zellmembranen
beteiligt ist, bestehen aus einer Fettsäure verknüpft
(verestert)
mit dem Alkohol Sphingosin
![]()
![]()
2) Symptome:
- Kirchroter Fleck in der Makula (Augenhintergrund bei über
95%
der Patienten)
- Zunehmende Muskelschwäche nach dem 3. Lebensmonat
- Schreckreaktion auf Schallreize
- Psychomotorischer Abbau, Verlust des Sitz und
Stehvermögens
- Schwerhörigkeit, Blindheit, Krämpfe, Paresen, Spastik
- puppenartiges Gesicht mit blasser durchscheinender Haut
- lange Augenwimpern und feines Haar
![]()
![]()
3) Vererbung:
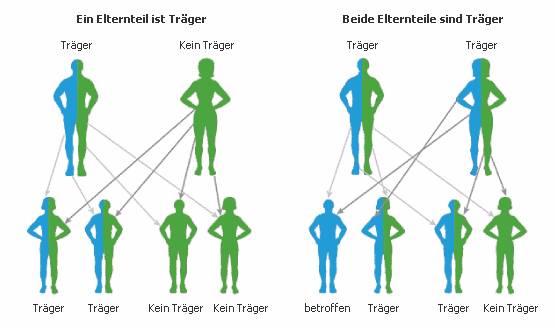
Die Tay-Sachs- Krankheit wird autosomal-rezessiv* vererbt, das bedeutet,
dass die Krankheit nur dann ausbricht, wenn ein Mensch zwei Kopien des
rezessiven Gens besitzt. Hat er dagegen nur eine Genkopie, bezeichnet
man ihn als „Träger“, d.h. er besitzt das „kranke“ Gen, ohne zu erkranken
(er weist keine Symptome auf).Bei der sexuellen
Fortpflanzung kommen
durch die Vereinigung der Gameten (Geschlechtszellen, Eizelle und Spermium)
zwei Chromosomensätze zusammen, je einer von jedem Elternteil. Dadurch
ist jedes Gen zweimal vorhanden. Die Ausprägung eines genetisch bestimmten
Merkmals (z. B. Haarfarbe) wird somit von Mutter und Vater gleichermaßen
beeinflusst. Die beiden Gene für ein bestimmtes Merkmal liegen auf den
homologen Chromosomen jeweils an der gleichen Stelle. Sind sie genetisch
völlig identisch, bezeichnet man die Person für dieses Gen als homozygot
(reinerbig). Wenn hingegen zwei verschiedene Allele (Varianten) des gleichen
Gens vorhanden sind, wird die Person als heterozygot (mischerbig) für
dieses Gen bezeichnet. Setzt sich eines von zwei verschiedenen Allelen
gegen das andere bei der Merkmalsausprägung durch, nennt man dieses Allel
dominant. Die unterdrückte Allelvariante wird als rezessiv bezeichnet, d.h.
ihr Merkmal tritt nicht in Erscheinung. Nur im reinerbigen Fall, wenn das
rezessive Allel im homozygoten Zustand vorliegt, kommt das rezessive
Merkmal zum Vorschein.
Man kann „gesunde“ Träger dieses
rezessiven („kranken“) Gens heutzutage
durch Blutuntersuchung herausfinden. Meist erfolgt dies aber in der
Regel erst, nachdem ein an Tay-Sachs erkranktes Kind geboren ist. Haben
beide Eltern ein „krankes“ und ein „gesundes“ Gen zu vererben, besteht
für eine Schwangerschaft ein 25%iges Risiko, dass das Kind betroffen ist.
Dies kann aber durch Untersuchung des Fruchtwassers festgestellt werden.
*Rezessiv:
bei
einer rezessiven Erkrankung müssen beide Allele eines
bestimmten Genes mutiert sein, damit sich der Krankheitsphänotyp ausprägt
*Autosomal:
Krankheit, die auf einer Anomalie
(Defekt) in einem oder
mehreren der 22 Autosomenpaare (Nichtgeschlechts-Chromosomen)
beruht
![]()
![]()
4) Verlauf:
Die Veränderungen im Nervengewebe führen
zunehmend
zu einem Verlust
schon erworbener Fähigkeiten nach dem 3. Lebensmonat, dazu gehören u.a.
Schreckreaktionen auf Schallreize, Verlust des Sitz- und Stehvermögens,
zunehmende Schwerhörigkeit und Blindheit, sowie vermehrtes Erbrechen
nach dem 16. Lebensmonat. Weiterhin kommt es zu einer Vergrößerung des
Kopfes nach dem 16 Lebensmonat, da es zu einer Vermehrung der zwischen
den Nervenzellen liegenden Fasern (Glia) kommt. Daraufhin treten vermehrt
Krampfanfälle auf, zunehmende Muskelschwäche bis hin zu Lähmungen
(Parese, Spastik). Die Kinder versterben in der Regel an wiederkehrenden
Lungenentzündungen (Pneumonien) zwischen dem 1. und 4. Lebensjahr.
![]()
![]()
5) Behandlung:
Leider gibt es für die Tay-Sachs- Krankheit bisher keinerlei Therapie.
Die betroffenen Kinder werden in der Regel im ersten Lebensjahr auffällig,
die Krankheit schreitet unaufhaltsam fort und die Lebenserwartung liegt bei
maximal 4 Jahren. Die Wissenschaftler suchen nach der biochemischen und
molekularbiologischen Ursache, woraus sich vielleicht in weiter Zukunft einmal
Behandlungsmethoden entwickeln lassen.
![]()
![]()
Vielen Dank, an die
Biochemikerin A.Behrendt für Ihre
Unterstützung dieser
Tay-Sachs Erklärung